bimm143_github
My classwork from BIMM143 S24
Machine Learning 1
Muhammad Tariq
Today we explore the use of different data presentation tools on R like PCA
First up kmean()
Demo of using kmean() function in base R. First make some data with a known structure.
tmp <- c( rnorm(30,-3), rnorm(30,3))
x <- cbind(x=tmp, y=rev(tmp))
plot(x)
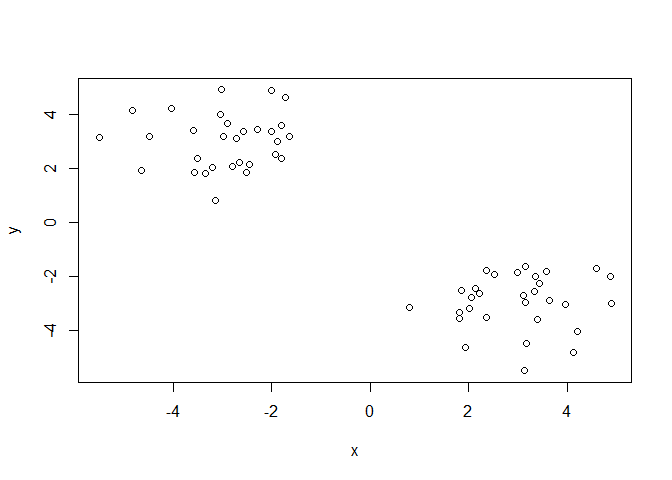
Now we have some made up data in ‘x’ lets see how kmeans() works with this data
k <- kmeans(x,centers = 2, nstart = 20)
k
K-means clustering with 2 clusters of sizes 30, 30
Cluster means:
x y
1 -2.948179 3.001639
2 3.001639 -2.948179
Clustering vector:
[1] 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 2 2 2 2 2 2 2 2
[39] 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2
Within cluster sum of squares by cluster:
[1] 57.94692 57.94692
(between_SS / total_SS = 90.2 %)
Available components:
[1] "cluster" "centers" "totss" "withinss" "tot.withinss"
[6] "betweenss" "size" "iter" "ifault"
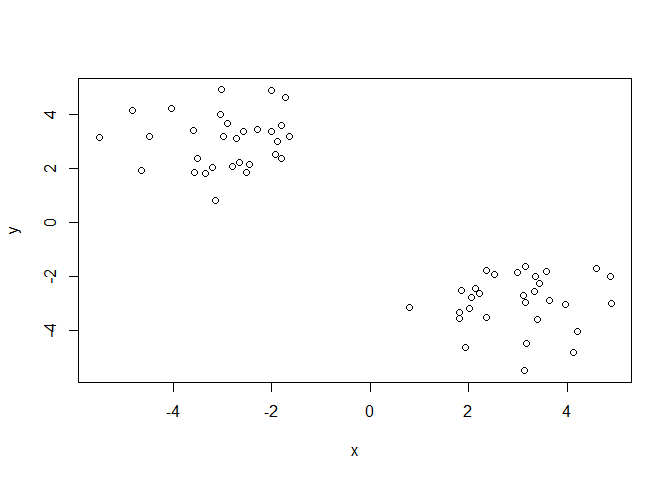
plot(x, col=k$cluster)
points(k$centers,col="blue",pch=15)
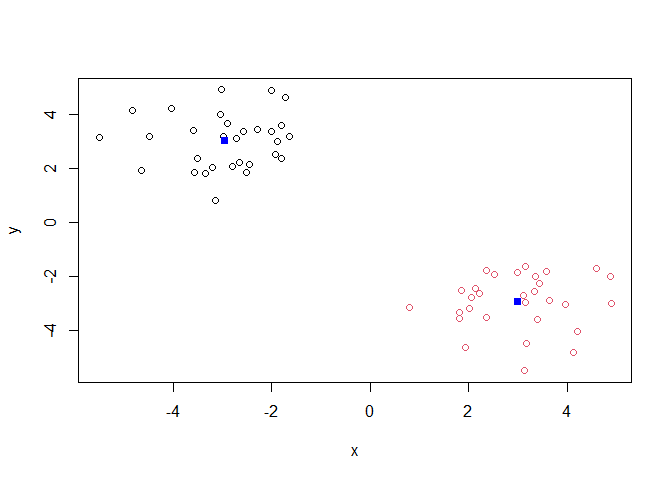
Add more clusters to the plots
k <- kmeans(x,centers = 4, nstart = 20)
plot(x, col=k$cluster)
points(k$centers,col="blue",pch=15)
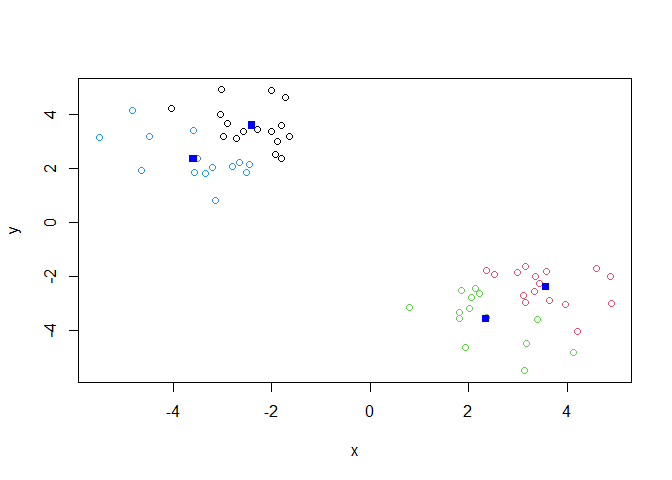
key-point; K-means clustering is supper popular but can be miss-used. one big limitation is that it can impose a cliustyering pattern on your data even if clear natural grouping dont exist- i.e it does what you tell it to do in therms of ‘center’.
Hierarchical Clustering
the main function in “base” R for hierarchical clustering is called ‘hclust’.
You can just pass our dataset as is into ‘hclust()’ you must give “distance matrix” as input. We can get this from the ‘dist()’ function in R.
d <- dist(x)
hc <- hclust(d)
hc
Call:
hclust(d = d)
Cluster method : complete
Distance : euclidean
Number of objects: 60
The results of ‘hclust)’ dont have a useful ‘print()” method but do have a speacial ’plot()’ method.
plot(hc)
abline(h=8, col="red")
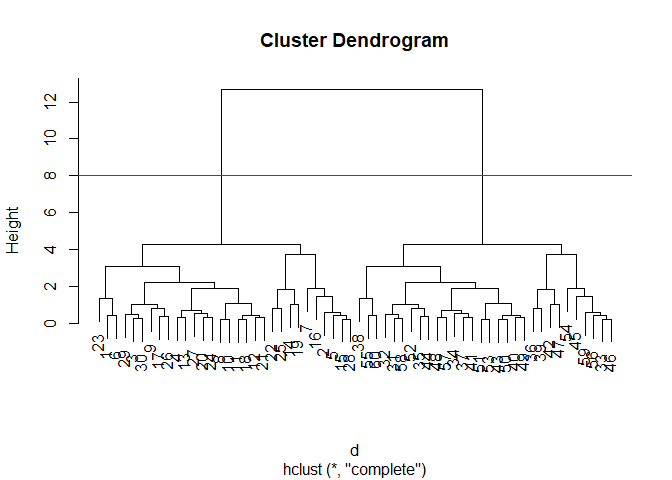
To get our main cluster assignmnet (membership vector) we need to “cut” the tree at the big goal posts
grps <- cutree(hc, h=8)
grps
[1] 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 2 2 2 2 2 2 2 2
[39] 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2
table(grps)
grps
1 2
30 30
plot(x)
Hierarchical clustering is distinct in that the dendrogram (tree figure) can reveal the potential grouping in your data (unlike K-means)
Principal Component Analysis (PCA)
PCA is a common and highly useful dimensionality reduction technique used in many fields - particularly bioinformatics.
Here we eill analyze some data from UK on food consumption.
###data import
url <- "https://tinyurl.com/UK-foods"
x <- read.csv(url)
head(x)
X England Wales Scotland N.Ireland
1 Cheese 105 103 103 66
2 Carcass_meat 245 227 242 267
3 Other_meat 685 803 750 586
4 Fish 147 160 122 93
5 Fats_and_oils 193 235 184 209
6 Sugars 156 175 147 139
rownames(x)<- x[,1]
x <- x[,-1]
head(x)
England Wales Scotland N.Ireland
Cheese 105 103 103 66
Carcass_meat 245 227 242 267
Other_meat 685 803 750 586
Fish 147 160 122 93
Fats_and_oils 193 235 184 209
Sugars 156 175 147 139
x <- read.csv(url, row.names=1)
head(x)
England Wales Scotland N.Ireland
Cheese 105 103 103 66
Carcass_meat 245 227 242 267
Other_meat 685 803 750 586
Fish 147 160 122 93
Fats_and_oils 193 235 184 209
Sugars 156 175 147 139
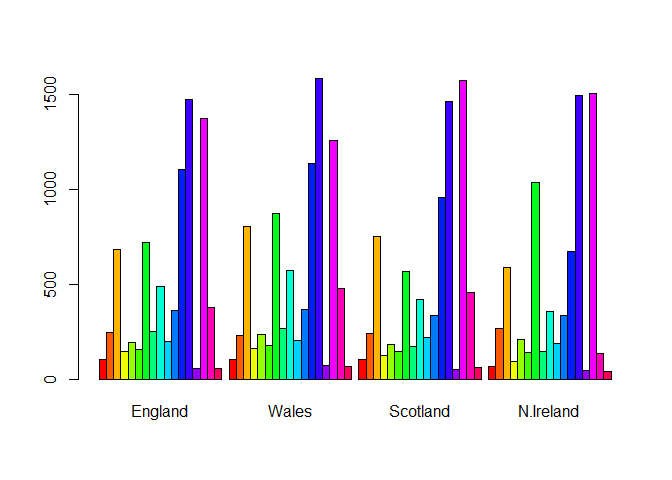
barplot(as.matrix(x), beside=T, col=rainbow(nrow(x)))
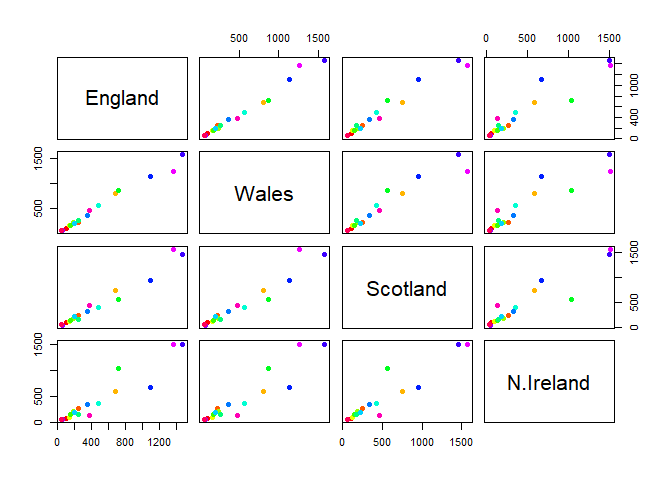
one conventional plot that can be useful is called a “paris” plot.
pairs(x, col=rainbow(nrow(x)), pch=16)
PCA TO THE RESUE
The main function in base R for PCA is called ‘prcomp()’
pca <- prcomp(t(x))
summary(pca)
Importance of components:
PC1 PC2 PC3 PC4
Standard deviation 324.1502 212.7478 73.87622 3.176e-14
Proportion of Variance 0.6744 0.2905 0.03503 0.000e+00
Cumulative Proportion 0.6744 0.9650 1.00000 1.000e+00
Interpretting PCA results
The ’prcomp() function returns a list object of our results with five attributes/components
attributes(pca)
$names
[1] "sdev" "rotation" "center" "scale" "x"
$class
[1] "prcomp"
The two main “results” in here are ‘pca$x’ and ‘pca$rotation’. the first of these (pca$x’) contains he scores of the data on the new PC axis - we use these to make our “PCA plot”.
pca$x
PC1 PC2 PC3 PC4
England -144.99315 -2.532999 105.768945 -4.894696e-14
Wales -240.52915 -224.646925 -56.475555 5.700024e-13
Scotland -91.86934 286.081786 -44.415495 -7.460785e-13
N.Ireland 477.39164 -58.901862 -4.877895 2.321303e-13
library(ggplot2)
#Make a plot of pca$x with PC1 vs PC2
ggplot(pca$x) +
aes(PC1, PC2, label=rownames(pca$x)) +
geom_point() +
geom_text()
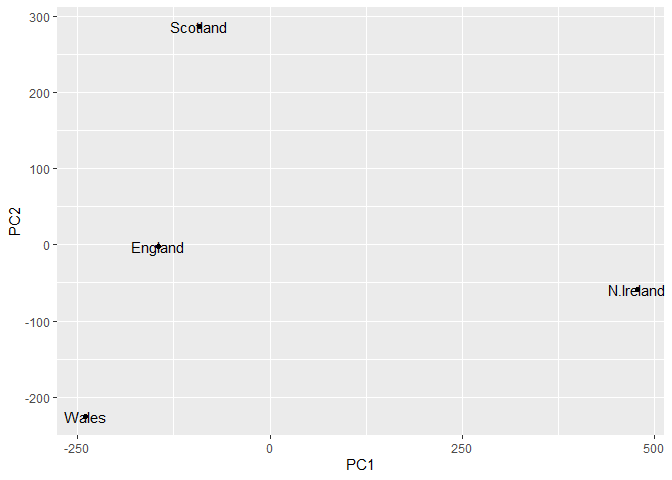
! PC1 enlightens the differnces in the rows, in how different the foods are consumed.
The second major result is contained in the ‘pca$rotation’ object or component. Let’s plot this to see what PCA is picking up…
ggplot(pca$rotation) +
aes(PC1, rownames(pca$rotation)) +
geom_col()
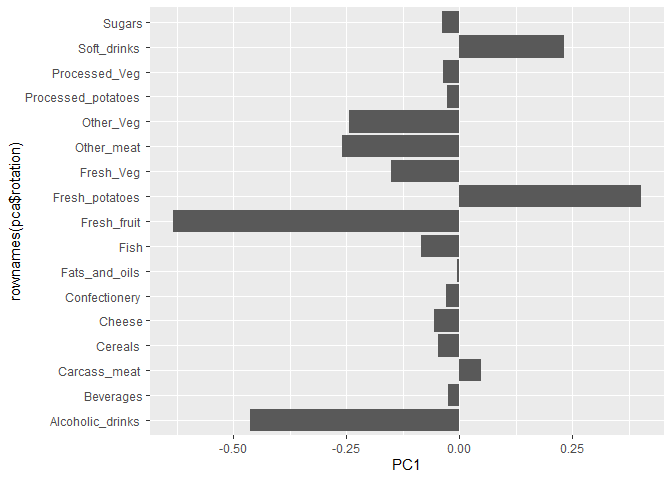
!it shows how which food is eaten so differently.